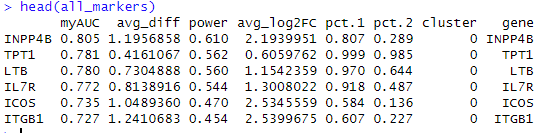
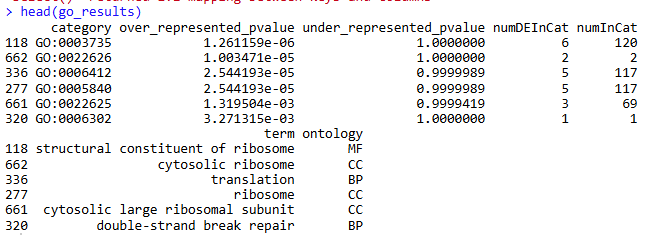
Bioinformatic analysis:linux操作指南之上游分析(Part 1)
· 6 min read

1. 安装 linux
这就不多说了,自己搞一个虚拟机,我用的是 Centos7。
ps:如果使用的是学校集群的话,注意在修改密码中改一下自己的密码,开启后账号为:root,密码自定义(注意是暗文,你敲进去是不会显示的)结束了 enter 即可
2. 预先安装
首先要安装 anaconda,为了不污染环境
-- 安装linux安装包(如果报错自己去anaconda网站找地址)
wget https://repo.anaconda.com/archive/Anaconda3-2023.07-Linux-x86_64.sh
-- 解压anaconda,后面的就是刚刚安装好的名字
bash Anaconda3-2023.07-Linux-x86_64.sh
-- 更新环境变量
source ~/.bashrc